da responsabilidade exclusiva dos seus autores.























-
Pneumologista
Serviço de Pneumologia do Centro Hospitalar e Universitário de Coimbra
PF-ILD significa Progressive Fibrosing Interstitial Lung Disease, ou em Português, DPI-FP, doença pulmonar intersticial fibrosante e progressiva.
O grupo das doenças pulmonares intersticiais (DPI) é extenso e heterogéneo, não existindo ainda uma classificação globalmente consensual das mesmas. Uma possibilidade é classificá-las em doenças de causa desconhecida, doenças de causa conhecida (autoimune sistémica ou exposição), granu-lomatoses e entidades raras. Cada uma das doenças específicas pode ser enquadrada num destes grupos.1 Na figura 1 pode encontrar-se uma proposta de classificação para as doenças mais comuns.
Figura 1: Proposta de classificação das doenças intersticiais mais comuns. São usadas as siglas habituais. DTC: doenças do tecido conjuntivo; PH: pneumonite de hipersensibilidade; PII: pneumonias intersticiais idiopáticas; LAM: linfangioleiomiomatose; PLCH: histiocitose pulmonar de células de Langerhans; FPI: fibrose pulmonar idiopática; NSIP: pneumonia intersticial não específica; RB-ILD: bronquiolite respiratória com doença pulmonar intersticial; DIP: pneumonia intersticial descamativa; COP: pneumonia em organização criptogénica; AIP: pneumonia intersticial aguda; LIP: pneumonia intersticial linfocítica; PPFE: fibroelastose pleuroparenquimatosa.

Um dos aspetos mais característicos das DPI é a importante variabilidade do seu comportamento evolutivo, com ou sem tratamento. Algumas destas doenças são exclusivamente inflamatórias, mas a maioria pode cursar com fibrose pulmonar, definida como uma desorganização arquitetural irreversível do parênquima pulmonar. Quando o processo de fibrose ocorre e progride apesar do tratamento adequado, podemos classificá-la como doença fibrosante progressiva. O con-ceito deriva de uma classificação alternativa das doenças de acordo com o seu comportamento clínico, inicialmente proposta por Athol Wells em 2003 e integrada na classificação das pneumonias intersticiais idiopáticas em 2013.2 Esta classificação do comportamento é utilizada para eleger a melhor estratégia de monitorização e tratamento de uma forma quase independente do diagnóstico específico. Na tabela 1, podemos encontrar a classificação de comportamento de doença publicada na classificação internacional das pneumonias intersticiais idiopáticas de 2013.
Tabela 1: Classificação de comportamento de doença, adaptado de Travis WD, et al.2

A DPI-FP corresponde à forma 5, progressão inexorável independentemente da terapêutica. Esta classificação corresponde ainda a uma versão juntar (lumping) da clássica discussão entre juntar ou separar (lumping vs. splitting) as doenças intersticiais.3
Refere-se que a junção das doenças neste fenótipo não reduz a importância de um diagnóstico preciso para cada doente, uma vez que a abordagem da doença progressiva não exclui o tratamento dirigido a uma entidade. Para uma discussão detalhada, consultar o último capítulo desta obra. Importa ainda mencionar que a definição de progressão tem sido alvo de discussão com criação de critérios que permitam classificar doentes quer para efeitos de tratamento quer para inclusão em ensaios clínicos. As várias propostas são descritas na pergunta 6: como é feita a avaliação da progressão nas DPI-FP?
O tratamento da DPI-FP é dependente do diagnóstico subjacente e deve incluir tanto medidas farmacológicas como não farmacológicas. A abordagem deve incluir sempre que possível a identificação e eliminação da exposição à causa da doença, especialmente na pneumonite de hipersensibilidade. Da mesma forma, o tabagismo deve ser abordado e a cessação tabágica implementada, tal como a vacinação antigripal e pneumocócica.
Em termos de medicação, o nintedanib foi aprovado pela FDA e pela EMA na FPI, na doença intersticial associada à esclerose sistémica e na DPI-FP, uma vez que reduz a progressão da doença.18-20 A pirfenidona foi aprovada na FPI pela EMA e FDA.21 A pirfenidona reduziu a progressão num ensaio clínico em doentes com doença inclassificável progressiva e mais de 10% de área de fibrose na TCAR. Apesar do endpoint primário deste ensaio não ter sido avaliável, os objetivos secundários permitem suportar a eficácia nesta população.14 Os corticoides ou outros imunossupressores são habitualmente prescritos quando há suspeita de substrato inflamatório, mas a evidência é escassa, exceto na sarcoidose e na esclerose sistémica. A escolha dos imunossupressores nas outras entidades é baseada em estudos de coorte e em recomendações de especialistas. As escolhas mais habituais para cada uns dos grupos de doentes podem ser encontradas na tabela 2.22
Nos doentes com limitação funcional, a reabilitação pulmonar tem bons resultados. Quando há hipoxemia de esforço ou em repouso pode ser prescrita oxigenoterapia. As comorbidades são frequentes e devem ser identificadas e tratadas. Nos casos mais graves e quando não há contraindicações, o doente deve ser precocemente referenciado para avaliação pré-transplante pulmonar. A terapêutica paliativa é um adjunto muito importante nos doentes com formas avançadas e pode ser instituída em paralelo com todas as estratégias já referidas.22
Tabela 2: Terapia imunossupressora e antifibrótica para as doenças intersticiais que podem decorrer com fenótipo DPI-FP. Adaptado de Wijsenbeek M, et al.22 MMF: micofenolato de mofetil.


-
Radiologista
Serviço de Radiologia do Centro Hospitalar Universitário de São João, EPE Porto
O diagnóstico de uma doença pulmonar fibrosante é feito em TCAR pela presença de distorção arquitetural, de opacidades reticulares (ou “padrão reticular”), de bronquiectasias e bronquiolectasias de tração e de padrão “em favo de mel”.3 Segundo a Fleischner Society7, a distorção arquitetural carateriza-se em TCAR por uma alteração da normal anatomia do lóbulo pulmonar secundário, geralmente associada a perda de volume pulmonar. O padrão reticular carateriza-se pela presença de inúmeras pequenas linhas dentro dos limites do lóbulo pulmonar secundário (simulando a presença de uma rede, daí o termo “reticular”) e correlaciona-se histologicamente com o espessamento dos septos inter e intralobulares. Este padrão não é específico das doenças fibróticas (pode surgir no edema pulmonar ou na linfangite carcinomatosa, por exemplo), mas geralmente acompanha as outras manifestações imagiológicas de fibrose pulmonar. As bronquiectasias (e bronquiolectasias) de tração traduzem uma dilatação irreversível e irregular das pequenas vias aéreas condicionada por um processo fibrosante circundante. O padrão “em favo de mel” (honeycombing) é considerado o sinal mais específico de fibrose pulmonar em TCAR e carateriza-se por um agrupamento de espaços císticos com diâmetros semelhantes (geralmente na ordem dos 3-10 mm), tipicamente em localização sub-pleural, com paredes bem delimitadas e, na maioria das vezes, partilhadas entre si (Figura 1).
Figura 1:
Padrão “em favo de mel”. Imagem axial de tomografia computorizada de alta resolução de doente com envolvimento pulmonar por artrite reumatoide demonstra um padrão reticular de predomínio periférico com padrão “em favo de mel” subpleural nos segmentos superiores dos lobos inferiores (setas).

Este sinal correlaciona-se histologicamente com fibrose pulmonar “terminal” (independentemente da causa). O padrão “em favo de mel” é um dos sinais fundamentais no algoritmo diagnóstico de pneumonia intersticial usual/ fibrose pulmonar idiopática (UIP/FPI).3-5 Note-se, contudo, que existe uma variabilidade inter e intraobservador considerável na avaliação destes achados imagiológicos (sobretudo na avaliação do padrão “em favo de mel”8), pelo que devem ser utilizados com precaução em relatórios radiológicos, tendo em conta as suas implicações diagnósticas e prognósticas. Faz-se, por último, uma referência ao padrão em “vidro despolido”.
Este padrão define-se como um aumento da opacidade do parênquima pulmonar com preservação das marcas vasculares e brônquicas subjacentes.7 Qualquer processo patológico que resulte na diminuição da quantidade de ar intra-alveolar (seja por preenchimento alveolar seja por espessa-mento do interstício pulmonar) traduz-se por opacidades em “vidro despolido” na TCAR. Este sinal acompanha muitas das doenças pulmonares fibrosantes difusas e, quando extenso, é importante no diagnóstico diferencial.3
São várias as doenças do interstício pulmonar que podem cursar com um fenótipo progressivo, destacando-se, pela sua frequência, as pneumonias intersticiais idiopáticas, as doenças autoimunes com envolvimento pulmonar, a pneumonite de hipersensibilidade, a sarcoidose e as doenças ocupacionais ou pneumoconioses2
Segundo a classificação conjunta da American Thoracic Society/European Respiratory Society (ATS/ERS)9, as pneumonias intersticiais idiopáticas que mais frequentemente tomam este curso clínico são a FPI e a pneumonia intersticial não-específica idiopática (iNSIP) - também chamadas de pneumonias intersticiais fibrosantes crónicas. O radiologista tem um papel fundamental no diagnóstico destas entidades em sede de reunião multidisciplinar. A caraterística fundamental da FPI é a presença de um padrão imagiológico ou histológico de UIP.3-5,9-11 Em TCAR, o padrão de UIP carateriza-se pela presença de um padrão reticular subpleural, de predomínio basal (inferior), com padrão “em favo de mel” e bronquiectasias ou bronquiolectasias de tração (Figura 2).3-5,12
Figura 2: Padrão de pneumonia intersticial usual típica na fibrose pulmonar idiopática. Imagens de tomografia computorizada de alta resolução demonstram perda de volume, distorção arquitetural e um padrão reticular periférico com bronquiectasias de tração e um extenso padrão “em favo de mel” com gradiente apicobasal.

No contexto clínico apropriado, quando todas estas caraterísticas estão presentes (o chamado padrão de “UIP típica”) e não há sinais imagiológicos sugestivos de um diagnóstico alternativo, diagnóstico de FPI pode ser feito sem recurso a biopsia pulmonar.4,5 Falamos de um padrão de “UIP provável” quando estamos perante um padrão reticular periférico de predomínio inferior, com bronquiectasias de tração, mas sem padrão “em favo de mel” inequívoco. Nestes casos, o diagnóstico mais provável continua a ser a FPI13, embora segundo a guideline conjunta da ATS/ERS seja necessária confirmação histológica nestes doentes.5 Por último, o padrão “indeterminado para UIP” foi introduzido apenas em 20184,5 e aplica-se quando existe um padrão reticular sem predomínio subpleural ou basal com alguns sinais imagiológicos menos frequentemente observados na FPI (fibrose com algum atingimento broncocêntrico ou opacidades em “vidro despolido” mais extensas do que o habitual, por exemplo). Nestes casos, a probabilidade de um diagnóstico de FPI vir a ser feito em reunião multidisciplinar ronda os 50%.3 Os achados imagiológicos em TCAR mais frequentes na pneumonia intersticial não- específica são as opacidades em “vidro despolido” bilaterais e de predomínio inferior, na maioria das vezes (75% dos casos) com espessamento septal intralobular associado (isto é, com reticulação), bronquiolectasias e bronquiectasias de tração.9,14,15 O padrão em “favo de mel” pode ocorrer, mas não é uma caraterística desta doença. Quando estas alterações “poupam” o parênquima pulmonar imediatamente sub-pleural (o chamado subpleural sparing), o diagnóstico de padrão pneumonia intersticial não-específica (NSIP) pode ser feito com mais confiança.14 Histologicamente, estas alterações traduzem a presença de inflamação e fibrose intersticial, com preservação da arquitetura pulmonar e com distribuição homogénea no tempo e no espaço2,9,15 (ao contrário da heterogeneidade temporal e espacial típica da UIP).
É preciso realçar, contudo, que quer o padrão UIP quer o padrão NSIP são padrões radiológicos e histológicos (e não doenças), podendo ser encontrados em muitos outros contextos clínicos (na pneumonite de hipersensibilidade fibrótica, no envolvimento pulmonar por conectivites, na asbestose ou na toxicidade farmacológica pulmonar, por exemplo). Esta afirmação é particularmente importante para o padrão NSIP, sendo muito mais frequentemente encontrado no contexto de uma doença de base (sendo o caso paradigmático as doenças do tecido conjuntivo) do que na forma idiopática16 (Figura 3).
Figura 3:
Pneumonia intersticial não-específica na esclerose sistémica. Padrão de opacificação em vidro despolido com reticulação intralobular associada e bronquiectasias de tração (setas), com predomínio inferior e distribuição relativamente homogénea. Nota-se ainda dilatação esofágica, habitual neste contexto (*).

As doenças do tecido conjuntivo (DTC) são um grupo heterogéneo de doenças inflamatórias sistémicas caraterizadas pela presença de autoanticorpos e lesão de órgão-alvo, sendo o pulmão um dos órgãos frequentemente envolvidos.2,17 Todos os componentes do tórax, nomeadamente as vias áereas, o interstício, a vasculatura pulmonar, a pleura e o pericárdio podem ser afetados nestas doenças.17-19 As DTC que mais frequentemente cursam com doença pulmonar difusa são a artrite reumatoide, a esclerose sistémica, o síndrome de Sjögren, as miosites (dermatomiosite/polimiosite), a doença mista do tecido conjuntivo e o lúpus eritematoso sistémico. Os padrões de envolvimento pulmonar pelas conectivites são histologicamente e radiologicamente semelhantes às suas equivalentes idiopáticas (padrão de UIP, NSIP, pneumonia organizativa, pneumonia intersticial linfocítica, bronquiolite obliterante, entre outras) e a sua frequência varia de acordo com a DTC subjacente19 (Tabela 1).
Tabela 2. Padrões de atingimento intersticial nas doenças do tecido conjuntivo
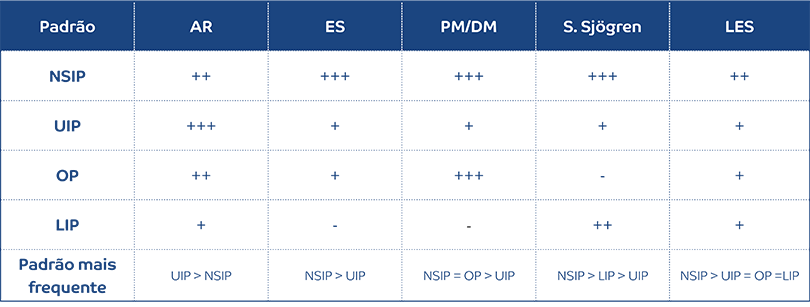
AR: artrite reumatoide; ES: esclerose sistémica; PM/DM: polimiosite/dermatomiosite; LES: lúpus eritematoso sistémico; NSIP: pneumonia intersticial não-específica; UIP: pneumonia intersticial usual; OP: pneumonia organizativa; LIP: pneumonia intersticial linfocítica. Adaptado de Ahuja J, et al. 2016.19
Regra geral, o padrão de NSIP é o mais frequentemente encontrado, exceto na artrite reumatoide onde o padrão de atingimento intersticial mais frequente é o padrão UIP.11,19 A coexistência de padrões (NSIP e pneumonia organizativa no contexto de uma miosite inflamatória, por exemplo) e o atingimento de vários compartimentos em simultâneo (coexistência de doença pulmonar intersticial com sinais de hipertensão pulmonar e derrame pleural/pleurite) é também uma caraterística do envolvimento pulmonar por conectivites. O radiologista deve também estar atento aos sinais extrapulmonares que possam ajudar no diagnóstico, nomeadamente: dilatação esofágica difusa, comum na esclerose sistémica; erosões das extremidades das clavículas na artrite reumatoide; calcificações dos tecidos moles na dermatomiosite; ou espessamento pleural e pericárdico (com ou sem derrame associado) no lúpus eritematoso sistémico.
A pneumonite de hipersensibilidade (PH) é uma doença complexa causada pela exposição continuada a um antigénio (mais comummente aviário, mas também microbiano ou químico), sendo a terceira causa mais frequente de doença pulmonar difusa (após a FPI e o envolvimento pulmonar pelas DTC).2,20 Os achados imagiológicos associados à pneumonite de hipersensibilidade refletem, na generalidade, a etiopatogenia da doença e traduzem-se por sinais de obstrução das pequenas vias aéreas (micronódulos centrilobulares, padrão de atenuação em mosaico ou air-trapping nos cortes obtidos em expiração) associadas a opacidades em “vidro despolido” (representando um processo inflamatório ou “infiltrativo”) com ou sem sinais de fibrose pulmonar. A recente guideline conjunta da American Thoracic Society/ Japanese Respiratory Society/Latin American Thoracic Association (ATS/ JRS/ALAT)6 para o diagnóstico de pneumonite de hipersensibilidade veio reclassificar esta doença nas categorias de PH não-fibrótica e PH fibrótica (por oposição à anterior classificação em PH aguda, subaguda e crónica). A este respeito, a coexistência de fibrose pulmonar e de sinais de obstrução bronquiolar é altamente sugestiva de PH fibrótica (Figura 4).
Figura 4:
Pneumonite de hipersensibilidade fibrótica. Opacidades reticulares irregulares com atingimento periférico e central (broncocêntrico), opacidades “em vidro despolido” e áreas lobulares de menor atenuação (setas) traduzindo obstrução das pequenas vias aéreas.

Os achados fibróticos na PH traduzem-se, na maioria das vezes, por um padrão reticular irregular com distorção arquitetural de distribuição difusa quer no plano axial quer no plano craniocaudal.6,20 A broncocentricidade dos achados fibróticos é também uma caraterística desta doença. A presença de bronquiectasias de tração ou de padrão “em favo de mel” é frequente, mas geralmente não constitui o padrão dominante. Um sinal imagiológico tido como específico de PH fibrótica é o “padrão das três densidades” (previamente chamado de padrão headcheese). Este sinal retrata a presença simultânea de três densidades pulmonares diferentes, coexistindo: (a) opacidades em vidro despolido (representando um processo inflamatório ou “infiltrativo”) e um padrão de atenuação em mosaico, constituído por (b) pulmão “normal” (de densidade intermédia), e (c) áreas lobulares de retenção aérea (de baixa densidade), traduzindo o processo obstrutivo das pequenas vias aéreas.6 Por último, salienta-se que, em alguns casos, a apresentação imagiológica pode ser virtualmente indistinguível da fibrose pulmonar idiopática (padrão de “UIP típica” ou “UIP provável”), da NSIP idiopática ou de uma DTC com atingimento pulmonar.

-
Anatomopatologista
Serviço de Anatomia Patológica do Centro Hospitalar Universitário de São João
Fibrose progressiva é uma característica importante de muitas doenças intersticiais e condiciona a morbilidade e a mortalidade. Apesar de muitas das doenças que se apresentam com fibrose progressiva terem aspetos clínicos, imagiológicos e patológicos distintos, tem também sobreposição de muitas características que são semelhantes à clássica doença intersticial pulmonar com fenótipo fibrosante, a Fibrose Pulmonar Idiopática (FPI).
A FPI, que é uma entidade
clinicopatológica distinta, é a forma mais comum de
fibrose pulmonar difusa e
progressiva.2,6
Caracteriza-se
histologicamente pelo padrão de Pneumonia
Intersticial Usual (UIP): heterogeneidade espacial e
temporal, áreas de fibrose densa, focos
fibroblásticos, áreas de parênquima poupado e áreas
de padrão em favo de mel (Figura 1).
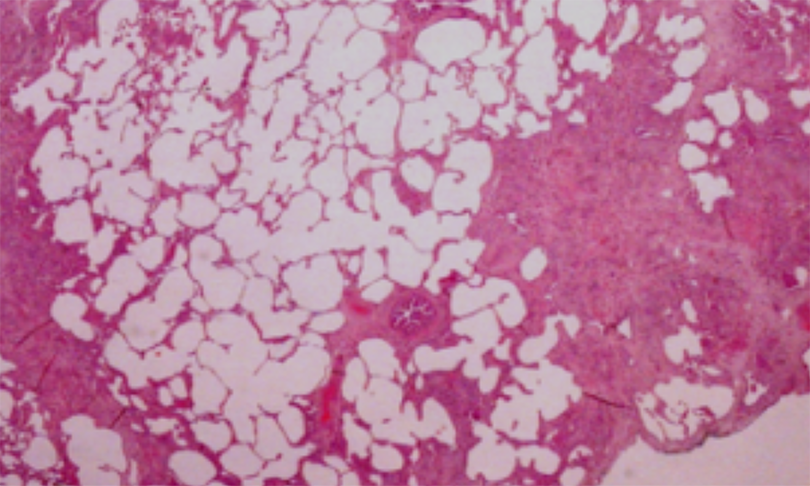
Figura 1: Biopsia cirúrgica HE40x - UIP. Áreas de fibrose densa alternando com parenquima pulmonar poupado.
A FPI é o arquétipo de uma doença fibrosante progressiva e pode ser vista como modelo das outras doenças intersticiais fibrosantes progressivas.2 Não é a única doença que leva a um end-stage de fibrose pulmonar. Várias outras pneumonias intersticiais podem ter este fenótipo fibrosante, como a pneumonia intersticial não especifica, pneumonia de hipersensibilidade, pneumonia intersticial associada a doenças do colagénio, sarcoidose e outras.
Doença pulmonar intersticial fibrosante progressiva foi redefinida como uma síndrome clinica que partilha características genéticas, patofisiológicas e história natural com a FPI.2
Pneumonite de hipersensibilidade - é uma
doença imunológica que se manifesta como doença
intersticial pulmonar em indivíduos suscetíveis, quando
expostos a um antígeno identificado ou não.3
Tem aspetos morfológicos que podem ser sobreponíveis a
outras pneumonias intersticiais. A pneumonite de
hipersensibilidade fibrótica difere da não fibrótica
porque para além da pneumonia intersticial crónica e/ou
bronquiolite é complicada de fibrose. A pneumonite de
hipersensibilidade fibrótica típica caracteriza-se por:
1 - pneumonia intersticial celular
centrada nas vias aéreas, assumindo por vezes um padrão
de NSIP celular e com predomínio de linfócitos.
2 - bronquiolite celular com predomínio
de linfócitos (linfócitos>plasmócitos), macrófagos
xantelasmizados nas vias aéreas terminais e pneumonia
organizativa.
3 - granulomas epitelioides malformados
não necrosantes.
4 - pneumonia fibrosante com distorção
arquitetural, focos fibroblásticos e favo de mel
sub-pleural; fibrose centrada nas vias aéreas com
metaplasia peri-bronquiolar (Figura 2).
Figura 2: Criobiopsia pulmonar transbrônquica HE 200x- Pneumonite de Hipersensibilidade - fibrose centolobular com metaplasia peribronquiolar.

Pneumonia intersticial não específica - histologicamente, a pneumonia intersticial não específica caracteriza-se por preservar a arquitetura do pulmão e envolver de forma uniforme o interstício.2 Observa-se infiltrado inflamatório intersticial mononuclear quase sempre associado a fibrose (Figura 3).
Figura 3: Criobiópsia pulmonar transbrônquica HE 200x - NSIP fibrose homogéneo do interstício pulmonar.

Pneumonia intersticial associada a doenças do
colagénio -
pneumonia intersticial ocorre em aproximadamente 15% dos
doentes com doença do colagénio, com maior incidência em
algumas patologias como esclerose sistémica e artrite
reumatoide.2
O padrão mais frequente de fibrose é de NSIP, mas um
padrão de UIP também pode ocorrer, particularmente na
artrite reumatoide, sendo que, neste caso, o diagnóstico
diferencial com FPI tem que ser feito. Alguns aspetos
morfológicos, como um infiltrado inflamatório
intersticial com predomínio de plasmócitos, pleurite e
coexistência de mais do que um padrão na biópsia,
favorecem envolvimento por doença do colagénio, bem como
a presença de bronquiolite folicular com centros
germinativos. Por outro lado, nestes casos existem
geralmente menos focos fibroblásticos do que na FPI
(Figura 4).
Figura 4: Criobiópsia pulmonar transbrônquica HE 100X: pleura com marcadas alterações reativas do mesotélio, fibrose e envolvimento por infiltrado inflamatório crónico com constituição de folículos linfoides, alguns com centro germinativo; parênquima pulmonar representado com espessamento homogéneo dos septos alveolares (NSIP fibrosante?).

No entanto, estas alterações podem ser discretas e têm que ser cuidadosamente avaliadas pelo patologista. O diagnóstico final é feito com base na integração dos aspetos histológicos com os fatores clínicos e imagiológicos.
Fibrose inclassificável - uma doença intersticial define-se como inclassificável quando os aspetos clínicos, imagiológicos ou histopatológicos são inadequados ou conflituosos entre si. Aproximadamente 10% das pneumonias intersticiais são inclassificáveis.5
Sarcoidose - é uma doença inflamatória rara, sistémica e de etiologia desconhecida. Resulta de uma resposta imunológica crónica a um antigénio desconhecido, em indivíduos geneticamente suscetíveis. Cerca de 90% dos doentes têm envolvimento pulmonar e cerca de 20% destes desenvolve fibrose (estádio IV).2 Caracteriza-se pela presença de granulomas epitelioides, bem formados e não necrosantes e, no estádio IV, fibrose que afeta predominantemente os segmentos posteriores dos lobos superiores e é centrada predominantemente nos eixos broncovasculares (Figura 5).
Figura 5: Criobiópsia pulmonar transbrônquica. HE 200x. Granulomas epitelioides bem formados, não necrosantes.

Doenças intersticiais relacionadas com exposição ocupacional - são causadas pela inalação e retenção no pulmão de várias partículas. As mais comuns são a abestose, causada pela inalação das fibras de asbestos e silicose relacionada com inalação de sílica.2 Na silicose simples observam- se pequenos nódulos de colagénio hialinizado, que ocorrem sobretudo nos lobos superiores. Nos casos avançados de silicose complicada, há confluência de nódulos, resultando em grandes massas com o desenvolvimento de fibrose progressiva predominantemente nos lobos superiores (Figura 6).
Figura 6: Criobiópsia pulmonar transbrônquica HE 200x - nódulo silicótico.

O padrão UIP é considerado um padrão associado a mau prognóstico. Nos últimos anos, muita literatura se tem referido a aspetos histológicos que podem ser marcadores de mau prognóstico e influenciar a sobrevida dos doentes com diferentes doenças fibrosantes pulmonares.4 Muita dessa literatura foca-se na importância dos focos fibroblásticos como manifestação de agressão pulmonar ativa. Em particular na FPI, a quantidade de focos fibroblasticos pode estar relacionada com o declínio da função respiratória e mortalidade. Os focos fibroblásticos correspondem a áreas de matriz mixoide com agregados de miofibroblastos ativos, produtores de colagénio (Figura 7) e integram os aspetos morfológicos típicos da FPI.
Figura 7: Biópsia cirúrgica HE 400X - Foco fibroblástico.

No entanto, podem estar presentes em outras pneumonias
fibrosantes, nomeadamente pneumonia intersticial não
específica e pneumonite de hipersensibilidade, embora
sejam geralmente menos numerosos.
Estudos recentes demonstraram que a quantidade de focos
fibroblásticos presentes na biópsia está relacionada com
a gravidade das bronquiectasias de tração identificadas
na TCAR.4 Este aspeto pode explicar a
evidência crescente de que a severidade das
bronquiectasias de tração é um fator prognóstico
importante nas doenças fibrosantes do pulmão.

-
Pneumologista
Serviço de Pneumologia, Centro Hospitalar Universitário de São João, Porto
Faculdade de Medicina da Universidade do Porto, Porto
-
Pneumologista
Centro Hospitalar Universitário de São João, Porto
Clinicamente, a FPI carateriza-se pelo desenvolvimento insidioso e progressivo de dispneia de esforço e/ou tosse, presença de crepitações inspiratórias bibasais na auscultação pulmonar e frequentemente hipocratismo digital. De salientar, a ausência de sintomas e sinais sugestivos de doença sistémica, nomeadamente de patologia autoimune, bem como exposição ambiental significativa ou toxicidade farmacológica. A exacerbação aguda da FPI, raramente, constitui a manifestação inicial da doença. Os doentes são maioritariamente do género masculino, com idade superior a 60 anos, e apresentam geralmente fatores de risco, como tabagismo, história familiar, ou refluxo gastroesofágico.
A tomografia computorizada com cortes de alta resolução (TCAR) é o método de diagnóstico central, cujo resultado define quatro categorias: padrão pneumonia intersticial usual (UIP); padrão UIP provável; indeterminado para UIP; e diagnóstico alternativo (Figura 1). 6 Num contexto clínico adequado, o padrão imagiológico de UIP “definitiva”, é patognomónico de FPI, com uma especificidade de 94-100%.7 Este padrão cursa com reticulação, bronquiectasias de tração e favo de mel de predomínio bibasal e subpleural, frequentemente num gradiente apicocaudal, na ausência de outros achados que possam sugerir um diagnóstico não-FPI, como áreas em mosaico, vidro despolido ou micronodulação.6 De salientar alguns achados radiológicos particulares: localização assimétrica (25%); ossificações nodulares (29%); associação com enfisema ou fibroelastose pleuropulmonar. Um padrão de UIP provável apresenta as mesmas alterações, exceto que não há evidência de favo de mel. Contudo, também num contexto clínico apropriado, este padrão assume relevância diagnóstica, correspondendo a cerca de 80% casos de UIP histológica. Por outro lado, só 30% dos casos de padrão indeterminado para UIP apresentam confirmação histológica de UIP.
Figura 1: Algoritmo diagnóstico para FPI. (a) Reticulação com predomínio subpleural e basal, de distribuição habitualmente heterogénea; alterações em favo de mel ± bonquiectasias de tração periféricas (b) Reticulação com predomínio subpleural e basal, de distribuição habitualmente heterogénea e com bonquiectasias de tração periféricas (sem favo de mel); pode ter discretas áreas de densidade de vidro despolido. (c) Reticulação subtil, sem óbvias caraterísticas de fibrose, ou áreas limitadas subpleurais/basais de opacificação em vidro despolido, que não sugere nenhuma etiologia específica, e que levanta a suspeita de UIP inicial.(d) Alterações sugestivas de outra etiologia: presença de cistos, marcada atenuação em mosaico, opacificação em vidro despolido dominante, consolidação, micronódulos dispersos, ou centrilobulares; distribuição predominante peribroncovascular, perilinfática, do andar superior ou médio do pulmão; presença de alterações pleurais (placas, espessamento, derrame), esófago dilatado, erosões claviculares distais, adenopatias relevantes. (e) Fibrose densa subpleural e parasseptal, com distorção da arquitetura pulmonar, alternando com áreas de pulmão não-afetado; presença de focos fibroblásticos e favo de mel.

Perante a necessidade de confirmação histológica do diagnóstico de FPI, e após a avaliação da relação risco/benefício, a biopsia pulmonar cirúrgica continua a ser o método mais consensual. 6 Este procedimento deverá ser efetuado, preferencialmente, por videotoracoscopia e com obtenção de múltiplas amostras referentes a dois ou mais lobos pulmonares, dada a possibilidade de coexistência de padrões histológicos distintos com implicações terapêuticas e prognósticas diferentes (por exemplo, UIP e pneumonia intersticial nãoespecífica [NSIP] fibrótica). A criobiopsia pulmonar transbrônquica é uma técnica endoscópica recente que permite a obtenção de amostras de tecido pulmonar de maior dimensão e qualidade do que a biopsia transbrônquica convencional.8 A sua acuidade diagnóstica é muito satisfatória, nomeadamente para os padrões de UIP e NSIP.
O padrão histológico de UIP, patognomónico de FPI, carateriza-se pela distorção da arquitetura do parênquima pulmonar, com fibrose colagenosa de predomínio subpleural e/ou parasseptal, favo de mel e focos fibroblásticos, com envolvimento heterogéneo, alternando com áreas de parênquima preservado (Figura 1). À semelhança do que foi descrito no âmbito da radiologia, também são reconhecidas quatro categorias histológicas: padrão UIP; UIP provável; indeterminado para UIP e diagnóstico alternativo.6

-
Reumatologistas
Serviço de Reumatologia do Hospital Garcia de Orta
-
Reumatologista
Serviço de Reumatologia do Centro Hospitalar e Universitário de Coimbra
-
Pneumologista
Serviço de Pneumologia do Centro Hospitalar e Universitário de Coimbra
Em todos os doentes com ES, devemos considerar medidas preventivas gerais de tratamento. Como quase todos estes doentes têm refluxo gastroesofágico, pelo envolvimento local da ES, devemos tratar esta condição (ex. inibidores da bomba de protões, anti-histamínicos H2, agentes procinéticos, adaptação do estilo de vida). É também recomendado vacinar estes doentes contra a gripe e contra a infeção por Pneumococos.
Num doente com ES e DPI, é controverso quando se deve iniciar tratamento e qual o melhor tratamento a usar. Regra geral, se o doente estiver sintomático ou houver sinais de progressão da doença, deve ser iniciado tratamento com um imunossupressor. Contudo, dada a modesta eficácia que estes fármacos podem ter e a toxicidade potencial, deve ser avaliado o risco/beneficio, tratando consoante o caso, sendo que os doentes mais graves e de maior risco devem sempre fazer tratamento. O tratamento deve ser iniciado precocemente, antes da evolução natural da doença para fibrose irreversível.
Doentes que apresentem nas PFV um declínio da FVC e/ou da DLCO, têm provavelmente doença ativa e devem iniciar tratamento. De igual modo, a presença de alterações na TCAR > 20% representam doença ativa, com maior risco de agravamento da doença e mortalidade.
A escolha do fármaco inicial tem recaído sobre o Micofenolato Mofetil (MMF), muito baseado nos resultados do Scleroderma Lung Study II, que compara este fármaco com a ciclofosfamida, revelando um bom perfil de eficácia e de segurança para o MMF.14
O MMF é utilizado geralmente numa dose entre 1,5 e 3g, dividido em 2 tomas, sendo os principais efeitos secundários alterações gastrointestinais e toxicidade medular. É um fármaco muito bem tolerado, mesmo por períodos prolongados de tempo, mostrando ser eficaz em melhorar os sintomas e os sinais de DPI, ou em estabilizar as alterações observadas.
A ciclofosfamida tem sido recomendada como 2ª linha de tratamento ou como alternativa ao MMF. A forma oral é adaptada ao peso (>= 2mg/Kg) e administrada durante 1 ano. No Scleroderma Lung Study I, mostrou ser eficaz com melhoria clínica e radiológica, bem como nas PFV com estabilização da FVC.15
A grande preocupação recai na toxicidade da ciclofosfamida, pelo que se tem dado preferência à toma endovenosa em vez da toma oral, sendo administrado IV mensalmente durante 6 meses, permitindo uma menor dose cumulativa e maior facilidade de hidratação. Esta via de administração provou ser também eficaz.16 Durante o tratamento deve-se monitorizar mensalmente o leucograma e a função renal (creatinina e sedimento urinário), para controlo de possíveis efeitos secundários. Contudo, cerca de um ano após a suspensão da ciclofosfamida, os benefícios observados desaparecem, pelo que se deve fazer uma terapêutica de manutenção com o MMF ou com azatioprina.
Os corticosteróides não devem ser utilizados nestes doentes, principalmente em doses altas, dado o risco de uma crise renal esclerodérmica, que é uma complicação muito grave e potencialmente fatal. Contudo, muitos dos doentes referidos nos estudos com os tratamentos anteriores, tomaram concomitantemente doses baixas de prednisolona (<10 mg/dia), sendo incerta a eficácia desta dose.
A azatioprina pode ser uma alternativa para doentes que não possam fazer MMF ou ciclofosfamida, mas parece ser menos eficaz como tratamento inicial da DPI.
Em doentes em que há progressão da doença, apesar do tratamento instituído, ou que não possam fazer MMF ou ciclofosfamida, o uso de Nintedanib pode ser uma boa opção. Este pode ser dado sozinho ou em associação com o MMF e demonstrou atrasar a progressão para fibrose no pulmão, diminuindo o declínio da FVC observada.17
O Rituximab foi também usado em doentes com DPI, mas os resultados observados são controversos, necessitando de mais estudos para se poder confirmar a sua eficácia nesta patologia. Contudo, em alguns estudos demonstrou ser eficaz, melhorando os valores de FVC e de DLCO, devendo ser considerado na doença refratária.
O Tocilizumab tem sido utilizado como tratamento alternativo, mas necessita de mais estudos para melhor definição de eficácia.
Outra opção a ter em conta é o transplante autólogo de células-tronco hematopoiéticas, muito eficaz na estabilização e mesmo regressão de lesões estabelecidas, mas com riscos importantes a indicar. Deve ser considerado quando há progressão da doença e risco de envolvimento de órgão.
Por fim, o transplante pulmonar pode ser considerado, para doentes muito selecionados com doença grave.
Figura 1: Sugestão de algoritmo de tratamento do envolvimento pulmonar intersticial na ES (adaptado de Systemic Sclerosis-associated interstitial lung disease. Lancet 2020; vol 8, Issue 3: 304:320)
TCAR: TAC de alta resolução; FVC: capacidade vital forçada; PFV: Provas de função ventilatória.


-
Pneumologistas
Serviço de Pneumologia Centro Hospitalar Vila Nova de Gaia/Espinho (CHVNG/E) Vila Nova de Gaia
Os agentes incitantes são diversificados e variam de acordo com a região geográfica. A exposição pode ser ocupacional, doméstica ou recreativa. Mais de 200 antigénios foram descritos para a PH e a cada ano novas causas são referidas.1 Dividem-se em agentes predominantemente orgânicos (bactérias, fungos, proteínas animais ou vegetais e enzimas) e inorgânicos (químicos de baixo peso molecular ou metais) (Tabela 1). 13
Em muitos casos, a exposição não é identificada. A latência entre a exposição e o início da doença pode variar de meses a décadas, constituindo um desafio para o clínico detetar o tipo e origem do antigénio, principalmente em casos de exposição desconhecida e em baixa quantidade.
Alguns autores sugerem que um agente incitante poderá fazer parte de uma mistura de microrganismos, proteínas e outras matérias (por exemplo, pó). Uma outra teoria é que determinantes antigénicos (epítopos) comuns poderão resultar em hipersensibilidade a múltiplos agentes incitantes.
Uma revisão recentemente publicada sobre exposições e associação com fenótipos clínicos, identificou os pássaros (32% dos casos de PH em estudos baseados em registos) e o bolor (17% dos casos) como as exposições mais frequentemente reportadas. Os estudos revistos descrevem taxas de nãoidentificação do Ag entre 15 a 24%. Os casos em que não se identificou o Ag, estavam mais vezes associados a um padrão fibrótico.14
Tabela 1. Causas mais comuns de PH de acordo com o tipo de exposição.
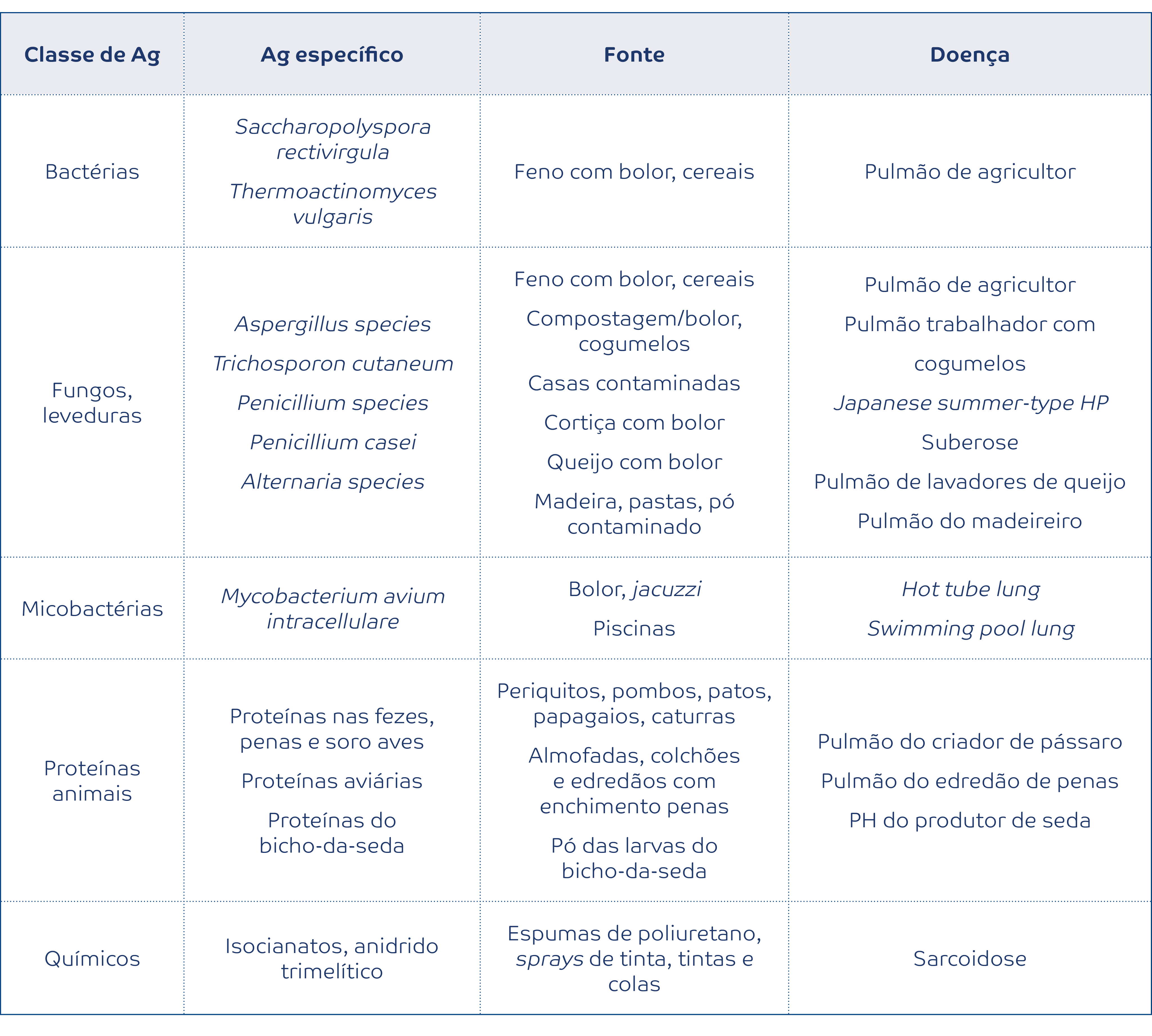
Adaptado de Spagnolo et al. J Investig Allergol Clin Immunol. 2015.
As caraterísticas radiológicas da PH traduzem o estadio histopatológico da doença. Desta forma, a mais recente guideline define as caraterísticas radiológicas atribuíveis a PH fibrótica e não-fibrótica, tendo em conta as alterações radiológicas descritas na literatura para cada subtipo e o impacto prognóstico das mesmas.1,16-23 Para cada um dos subtipos de PH, são estabelecidos critérios para os achados altamente sugestivos de PH (típico de PH), achados menos frequentemente reportados mas compatíveis com PH (compatível com PH) ou achados não sugestivos nem compatíveis com PH (indeterminado para PH). Estas caraterísticas encontram-se resumidas na tabela 2.
Tabela 2. Caraterísticas radiológicas (TCAR do tórax) do padrão de PH fibrótica.
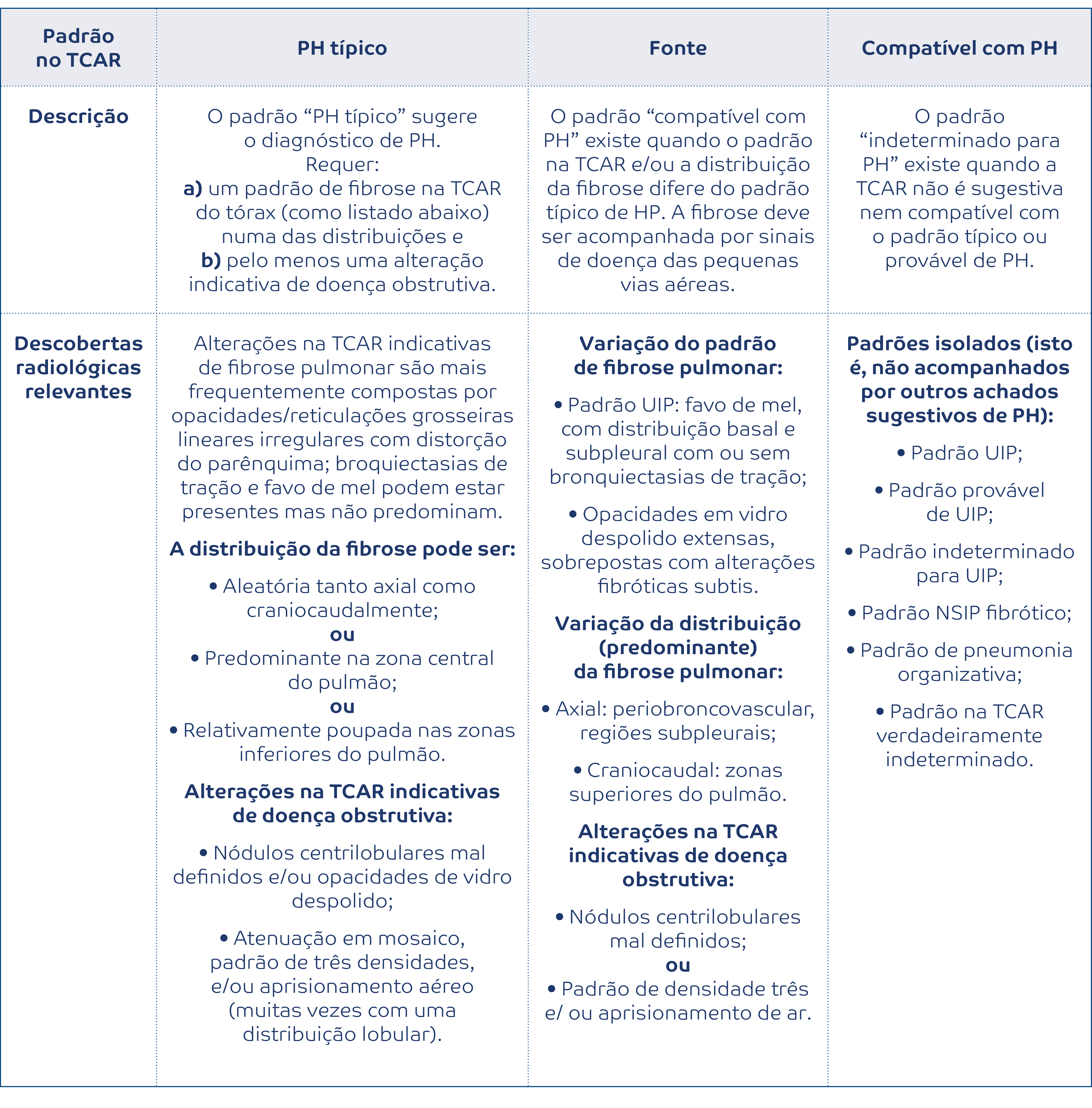
Caraterísticas radiológicas (TCAR do tórax) do padrão de PH fibrótica.
As caraterísticas histopatológicas típicas de PH
não-fibróticas consistem
em: bronquiolite crónica celular; infiltrado
inflamatório celular intersticial
bronquiolocêntrico; granunomas malformados e ausência de
caraterísticas
histopatológicas que sugiram um diagnóstico alternativo.
No entanto, nem
sempre todas as caraterísticas estão presentes. Na PH
fibrótica, à pneumonia
intersticial crónica subjacente acresce um padrão de
fibrose, por vezes
indistinguível das outras pneumonias intersticiais
idiopáticas como a fibrose
pulmonar idiopática ou a NSIP. Nestas situações, aspetos
como a polarização
brônquica das alterações histológicas ou fibrose em
ponte podem sugerir um
quadro de PH fibrótica.
A combinação das diferentes caraterísticas histológicas
observadas, tanto
para a PH não-fibrótica como para a PH fibrótica,
coloca-nos num cenário
histopatológico de PH definitiva, PH provável ou PH
indeterminada, de acordo
com a tabela 3.
Tabela 3. Critérios histopatológicos para o diagnóstico de PH (exceto Hot-Tub Lung*)

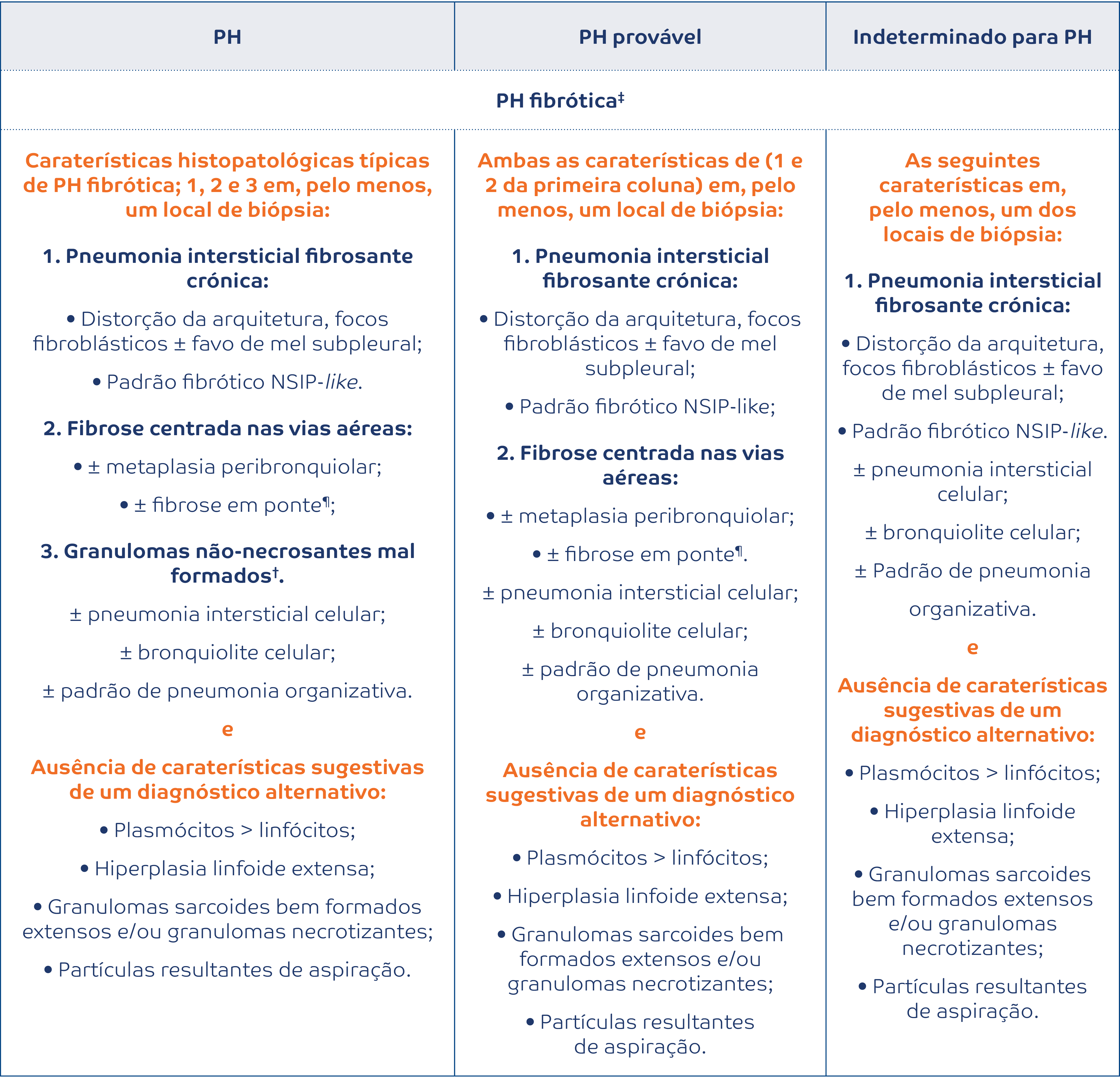
*Os achados histológicos da hot-tub lung são distintos das formas clássicas de PH fibrótica e não-fibrótica. † Os granulomas em PH são mais pequenos, mais laxos e não possuem a fibrose hialina perigranulomatosa, caraterística da sarcoidose. ‡ A PH fibrótica pode evidenciar as caraterísticas clássicas de PH não-fibrótica (PH celular) em áreas menos fibróticas ou não fibróticas; se presente, esta combinação de achados aponta para o diagnóstico de PH. ¶ fibrose em ponte estende-se da região subpleural à centriacinar ou liga focos fibróticos centriacinares vizinhos.
PH: pneumonite de hipersensibilidade; PII: Pneumonia intersticial idiopática; NSIP: pneumonia intersticial não-específica; UIP: pneumonia intersticial usual.
O prognóstico da PH é bastante heterogéneo, havendo doentes que evoluem para fibrose progressiva, falência respiratória e morte, com subgrupos a presentarem sobrevidas semelhantes à FPI.49,50 Numa publicação recente foi identificada uma série fatores associados ou preditores de sobrevida: caraterísticas demográficas; hábitos tabágicos; identificação e caraterísticas da exposição ao Ag; comorbilidades e marcadores fisiológicos, radiológicos e laboratoriais (Figura 1).50
Alguns aspetos estão consistentemente associados a uma maior mortalidade, como idade mais avançada, repercussão funcional mais severa à data do diagnóstico. Sexo masculino e Ag desconhecido parecem estarem também associados a uma maior mortalidade, mas os estudos não são consensuais.26,50 A presença de comorbilidades como hipertensão pulmonar, doenças autoimunes, fibroelastose pleuropulmonar associam-se a um pior prognóstico.50 A presença de fibrose (radiológica/histológica) está associada a maior mortalidade, particularmente na presença de favo de mel, maior extensão da doença e um maior volume vascular pulmonar. Salisbury avaliou um grupo de doentes com PH, de acordo com o fenótipo radiológico: o grupo de doentes sem evidência de fibrose apresentou uma melhoria progressiva da FVC, com sobrevida mediana livre de eventos > 14,73 anos; o grupo com fibrose sem favo apresentou uma sobrevida mediana de 7,95 anos; e o grupo com favo de mel apresentou uma sobrevida semelhante ao de uma corte emparelhada de doentes com FPI: 2,76 anos vs. 2,81 anos. A presença de polimorfismos do MUC5B e o encurtamento dos telómeros também são preditores de progressão.50
Figura 1: Sumário de fatores associados a aumento da mortalidade (adaptado de Creamer et al. Eur Respir Rev. 2020). Ag: antigénio; FVC: capacidade vital forçada; DLCO capacidade de difusão do CO; PH: pneumonite de hipersensibilidade; VVP: volume vascular pulmonar; UIP: pneumonia intersticial usual; NSIP: pneumonia intersticial não-específica.


Não Específica idiopática
-
Pneumologistas
Serviço de Pneumologia do Hospital da Luz Lisboa/ Hospital Beatriz Ângelo
A iNSIP tem uma taxa de sobrevivência de 80% aos 5 anos e de 73% aos 10 anos4, podendo utilizar-se o modelo GAP modificado ou ILD-GAP (Gender, Age, Physiology) para estimar a sobrevivência.33
No entanto, a história natural da iNSIP é imprevisível. O seu prognóstico é geralmente melhor do que o da FPI e inclui diversas possibilidades: alguns doentes melhoram, outros estabilizam e outros, ainda, agravam, com ou sem terapêutica.5
A extensão da fibrose tem impacto no prognóstico, parecendo haver diferenças na taxa de sobrevivência entre a NSIP celular e NSIP fibrótica, a favor da primeira.4,14
O padrão histológico basal é o melhor preditor de mortalidade das PII, mas os dados fisiológicos também fornecem informação relevante. Nos doentes com NSIP fibrótica e com DLCO (difusão do monóxido de carbono) inferior a 35% do previsto, o seu prognóstico e o da FPI tendem a ser semelhantes.34
Ainda assim, a mudança da função respiratória na evolução da doença parece ser mais importante do que a própria informação histológica.35
A avaliação da capacidade de exercício pode fornecer elementos prognósticos e facilitar decisões terapêuticas. Num estudo prospetivo que incluiu doentes com FPI e NSIP fibrótica, a dessaturação para valores inferiores a 88% na prova de marcha de 6 minutos associou-se a maior mortalidade.36
Desta forma, as características de pior prognóstico norteiam a referenciação do doente para transplante pulmonar: presença de NSIP fibrótica (independentemente da função respiratória); FVC inferior a 80% do previsto ou DLCO inferior a 40% do previsto; necessidade de oxigenoterapia, mesmo que apenas durante o esforço; resposta desfavorável ao tratamento imunossupressor, apresentando doença progressiva.34,37
Na avaliação da resposta ao tratamento e no follow-up dos doentes com iNSIP, apresenta-se uma proposta de monitorização, a adequar individualmente, e que sustenta a sua discussão dinâmica em reunião multidisciplinar (Tabela 3). 14
Tabela 3.Monitorização da iNSIP (adaptado de 14)
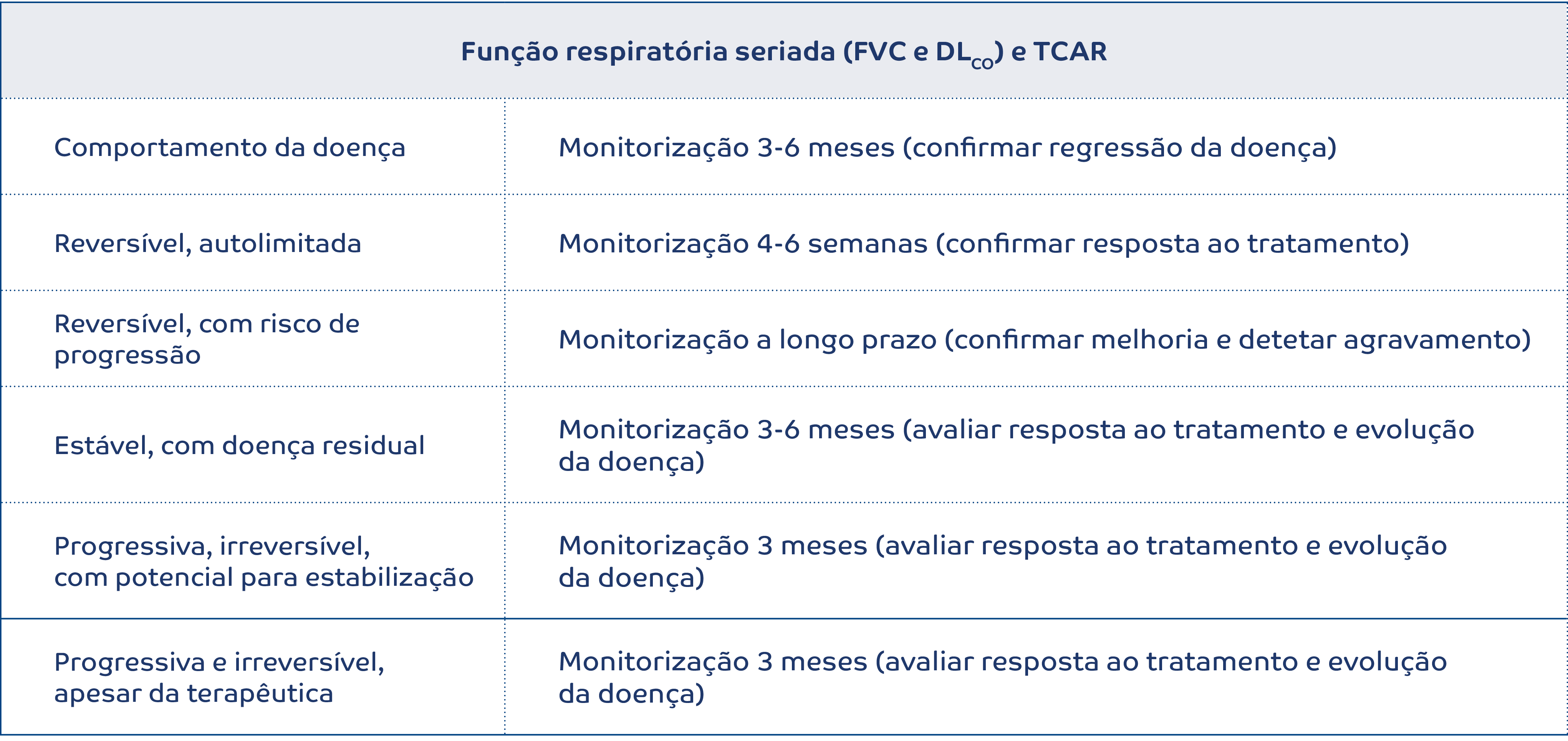
O acompanhamento destes doentes implica, para além da avaliação da função respiratória (FVC e DLCO) e imagiológica (TCAR), a monitorização de sintomas, da capacidade de exercício, de eventuais efeitos secundários da terapêutica e de comorbilidades.

-
Pneumologista
Serviço de Pneumologia Departamento Coração, Vasos e Tórax Hospital de Santa Marta Centro Hospitalar Universitário Lisboa Central
De acordo com o consenso de 2002 da ATS / ERS define-se como pneumonia intersticial idiopática inclassificável (inPII) uma pneumonia intersticial idiopática que, apesar de uma extensa avaliação multidisciplinar (clínica, radiológica e histológica), não se consegue enquadrar em nenhuma outra categoria.
Esta incapacidade de classificação pode resultar de diversos factores:
a) Impossibilidade de obter informação
essencial para prosseguir avaliação (ex.
ausência de condições para biópsia pulmonar, material da
biópsia insuficiente
para diagnóstico ou recusa por parte do doente);
b) Discrepâncias major entre os dados
clínicos, radiológicos e histológicos;
c) Terapêuticas prévias que
alteram/dificultam a interpretação dos achados
radiológicos ou histológicos;
d) Discrepância nos achados
histológicos em diferentes amostras, que não
consegue ser resolvida após integração com os dados
clínicos e radiológicos.
É importante ressalvar que todas estas situações devem
ser abordadas de
forma multidisciplinar e devem ser envidados todos os
esforços para tentar
identificar um diagnóstico.
Paralelamente, alguns autores propuseram dividir esta categoria em dois grupos:3
• um grupo em que foi obtida informação histológica
adequada e de qualidade
aceitável, que denominam de inclassificável;
• um segundo grupo em que não foi obtida biópsia ou o
material da mesma não
foi suficiente para uma análise satisfatória, que
denominam de não classificado.
Mais recentemente foi proposto um algoritmo diagnóstico, no qual, após integração da informação clínica, laboratorial (incluído estudo marcadores serológicos de autoimunidade), radiológica e histológica, se atribui um grau de probabilidade / confiança ao diagnóstico, denominando-se de inclassificável as situações em que o grau de confiança no diagnóstico é inferior a 50%.4
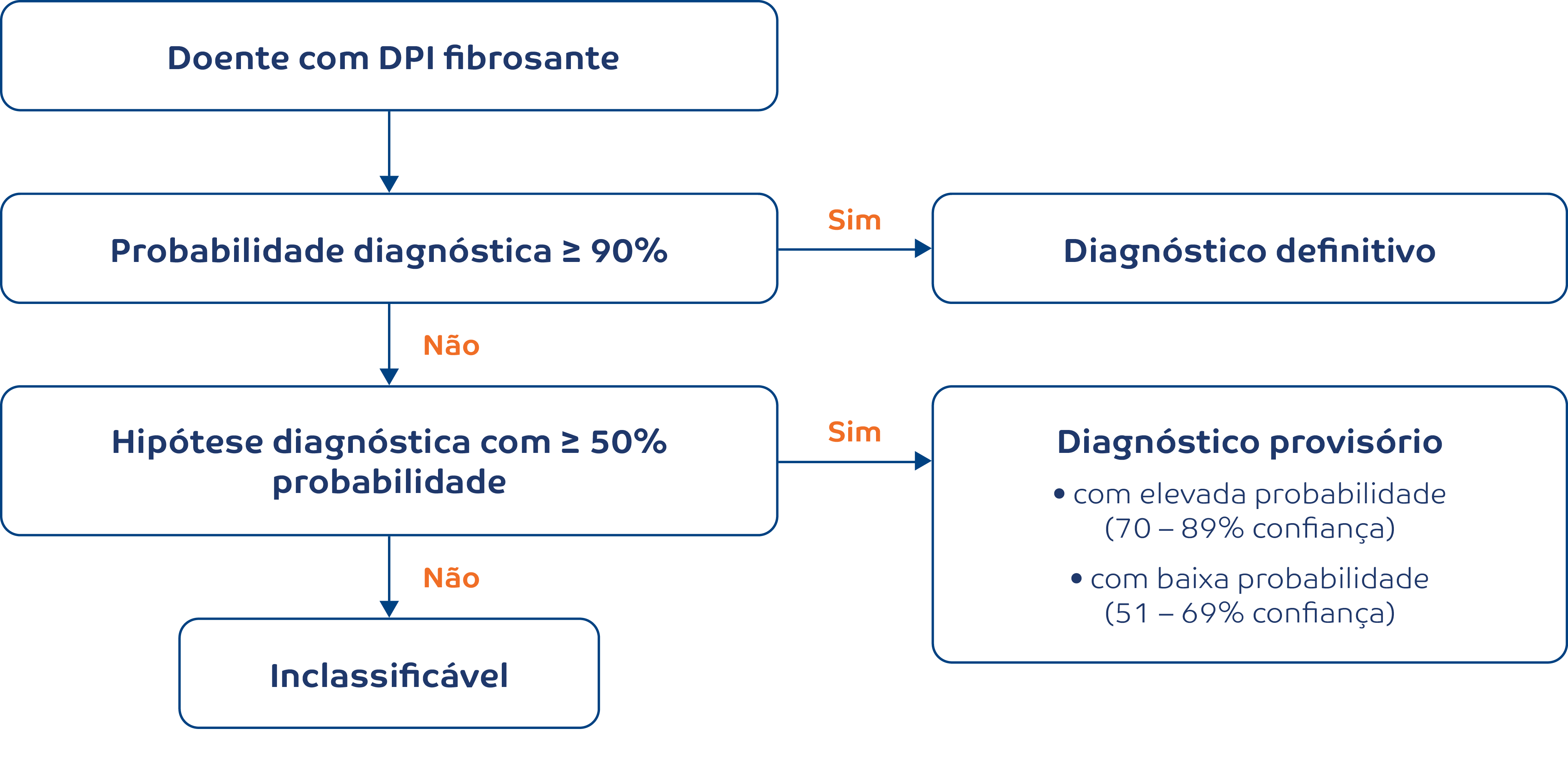
Figura 1: Proposta de classificação com base na probabilidade diagnóstica (adaptado de 4).
As opções terapêuticas destes doentes incluem, como habitual, um componente farmacológico e um componente não farmacológico. Em relação ao tratamento farmacológico não existem muitos estudos que visem especificamente esta população, pelo que as opções terapêuticas devem ser definidas caso a caso. A abordagem mais utilizada tem sido definir, em reunião multidisciplinar, quais os principais diagnósticos diferenciais e, dentro destes, qual o diagnóstico mais provável, prosseguindo para o tratamento farmacológico mais adequado para esse diagnóstico. Neste tipo de abordagem, o passo essencial é definir se o diagnóstico mais provável é ou não Fibrose Pulmonar Idiopática.
Em alternativa, alguns autores propõem uma abordagem mais abrangente, focada no comportamento da doença, que se encontra resumida na figura 2.4
Em relação ao tratamento não farmacológico, tal como a todos os doentes com patologia intersticial, também a estes doentes se recomenda cessação tabágica, vacinação antipneumocócica e antigripal, reabilitação respiratória, tratamento das comorbilidades e, quando indicado, oxigenoterapia de longa duração e transplantação pulmonar.
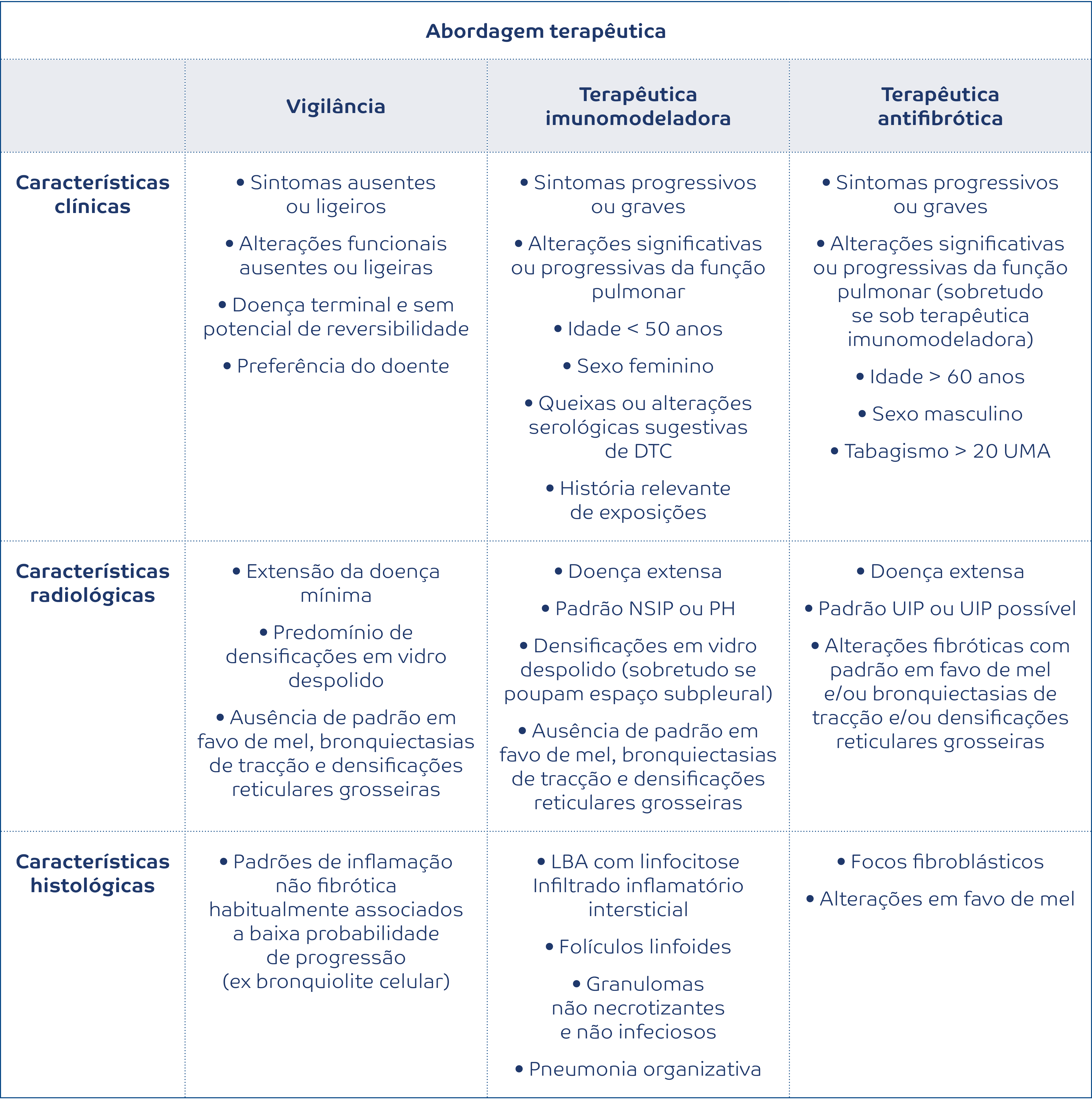
Figura 2: Proposta de abordagem terapêutica na Pneumonia Intersticial Inclassificável (adaptado de 4).
DTC: doença do tecido conjuntivo; LBA: lavado broncoalveolar; NSIP: pneumonia intersticial não específica; PH: pneumonite de hipersensibilidade; UIP: pneumonia intersticial usual, UMA: Unidade-maço / ano.






